Home › What is SENDIG v3.1.1 from CDISC?
All pharmaceutical companies, biotechs and contract research organizations (CROs) are required to prepare data according to the CDISC formats. Sponsors will be required to use these formats by the FDA, and consequently, CROs are increasingly expected to provide study results in these formats. Working with commercial software vendors can help ease the burden of complying with evolving format standards, while also helping you leverage best practices in a GLP regulated environment.
All pharmaceutical companies, biotechs, and contract research organizations (CROs) involved in preparing nonclinical data for submission to the FDA are required to prepare data according to the CDISC SENDIG v3.1.1 (Standard for Exchange of Nonclinical Data) format. Whether you are a sponsor company or a CRO, you are required to conform to a consistent format and terminology, which will streamline inter-organizational data exchange and generally enable the FDA to compare and analyze study information, potentially accelerating the approval process and throughput, In addition to the new data format, you need to ensure that your processes continue to meet Good Laboratory Practice (GLP) and CFR-21 Part 11 regulations and that you have the documentation to prove it.
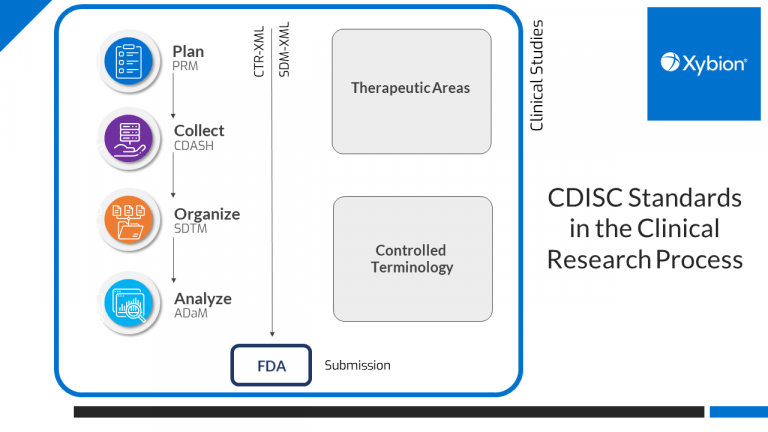
The data that makes up these submissions must be collected following the already established GLP and 21 CFR-Part 11 regulations. The data submitted in the SENDIG v3.1.1 format must accurately represent the study setup and observations, and laboratories are required to ensure this is the case. The SENDIG v3.1.1 submission requires precise documentation of dates, times and methods followed. For this reason, it is more important than ever that your processes support these regulations and that you have the validation framework in place to prove that your processes are being followed.

For NDA, ANDA, and certain BLA submissions, studies which start after December 18th, 2016, must have their data electronic submitted following the CDISC SENDIG v3.1.1 standards. For commercial INDs and amendments, except for submissions described in section 561 of the Federal Food, Drug, and Cosmetic Act, this is true for studies that started last December 18th, 2017. Most companies are interpreting this as a need to be SENDIG v3.1.1 ready before the end of 2015.
Savante facilitates the consolidation and formatting of non-clinical data and is designed to produce SENDIG v3.1.1 compliant data submissions to the FDA from a variety of data sources (including but not limited to Xybion’s Pristima Web pre-clinical data collection system).
Xybion QMS is a compliance and quality management solution that works in conjunction with Savante – the company’s state-of-the-art data warehousing and SENDIG v3.1.1 data preparation tool. It is used to organize and manage your SOPs and electronic content including SENDIG v3.1.1 datasets and non-clinical study reports. It has modules to create a master schedule of studies, approval workflows, and to ensure you are following GLP regulations through documentation, training, and internal audits.
We use cookies to improve your experience. By continuing to use our site, you accept our use of cookies. Privacy Policy and Terms of Use.
Cookie SettingsAccept